 |
Research Article
Mendelian randomization analysis of the causal association between immune cells and pancreatic cancer
1 Department of Radiation Oncology, Oncology Center, Zhujiang Hospital of Southern Medical University, No. 253 Mid Gongye Ave, Haizhu District, Guangzhou, Guangdong Province, Postal code 510282, China
2 Department of Radiation Oncology, Luang Me Hospital of University of Health Sciences, Street 271, Phnom Penh, Postal code 120110, Cambodia
Address correspondence to:
Duanyu Wang
and Jiqiang Li
Department of Radiation Oncology, Oncology Center, Zhujiang Hospital of Southern Medical University, No. 253 Mid Gongye Ave, Haizhu District, Guangzhou, Guangdong Province, Postal code 510282,
China
Message to Corresponding Author
Article ID: 100019G01PN2025
Access full text article on other devices

Access PDF of article on other devices

How to cite this article
Nov P, Touch S, Sou S, Kouy S, Wang D, Li J. Mendelian randomization analysis of the causal association between immune cells and pancreatic cancer. Edorium J Gastroenterol 2025;10(1):1–10.ABSTRACT
Aims: Pancreatic cancer (PC) is a highly lethal malignancy with limited treatment options. Tumor-infiltrating immune cells have been implicated in the progression and prognosis of PC. However, the causal role of immune cell populations in pancreatic cancer development and progression remains unclear. This study aims to elucidate the causal relationships between specific immune cell populations and the risk of pancreatic cancer, addressing gaps in current understanding.
Method: We conducted an extensive two-sample Mendelian randomization (MR) analysis. Using publicly available genetic data, we investigated the causal relationship between 731 immune cells and PC. We used inverse variance weighting (IVW) and weighted medians for MR analyses and used sensitivity analyses to assess heterogeneity and pleiotropy.
Results: In terms of the association between immune cells and PC, we found that CD62L- HLA DR++ monocyte % monocytes (OR = 1.1081, 95% CI = 1.0175–1.2068, p = 0.0184), SSC–A on HLA DR+ CD8br (OR = 1.1068, 95% CI = 1.0024-1.2221, p = 0.0448), CD64 on monocytes (OR = 0.8594, 95% CI = 0.8021-0.9207, p < 0.001), double-negative (DN) (CD4-CD8-) NKT %T cells (OR = 0.8712, 95% CI = 0.7802-0.9728, p = 0.0143), and SSC–A on HLA DR+ CD4+ cells (OR = 0.8902, 95% CI = 0.8028-0.9870, p = 0.0272) were strongly associated with PC. Among them, CD62L- HLA DR++ monocyte % monocytes and SSC–A on HLA DR+ CD8br are the risk factors, while CD64 on monocytes, DN (CD4-CD8-) NKT %T cells, and SSC–A on HLA DR+ CD4+ cells are protective factors for PC.
Conclusion: Our analysis provides evidence for a causal relationship between specific immune cell populations and PC. Targeting immune cell populations with therapeutic interventions such as immunotherapies may hold promise for improving outcomes in PC patients. Further studies are warranted to validate these findings and explore the underlying mechanisms involved in the immune response to PC.
Keywords: Genome wide association study (GWAS), Immune cells, Mendelian randomization (MR), Pancreatic cancer
INTRODUCTION
Pancreatic cancer (PC), a leading cause of cancer-related deaths, is anticipated to become the second leading cause of cancer mortality in the United States in the coming decades due to its high fatality rate. The 5-year survival rate is approximately 10%, as most patients (80–85%) present with either advanced or metastatic disease at diagnosis [1],[2]. In 2019, the American Cancer Society reported about 56,000 new cases and an estimated 45,000 deaths from PC, placing it behind lung and colorectal cancer [1]. Globally, it ranks seventh in cancer deaths, with GLOBOCAN 2018 estimating about 459,000 new cases and 432,000 deaths [3]. Pancreatic cancer is projected to surpass breast cancer as the third leading cause of cancer death in the European Union (EU) [3]. The high lethality is primarily due to late detection, often post-distant metastasis, and the absence of a single risk factor in most cases not associated with known factors or genetic mutations. Several factors have been suggested as reasons behind the observed increase in incidence rates. These include high rates of tobacco smoking, obesity, diabetes mellitus, lack of physical activity, and the consumption of high-fat/high-calorie diets, particularly prevalent in specific countries [4],[5],[6],[7]. Alongside these lifestyle factors, enhancements in the medical identification and diagnosis of PC, as well as a rising average life expectancy worldwide, also contribute to the increased incidence [8],[9].
Pancreatic cancer (PC) is frequently described as a “cold tumor,” [10],[11] characterized by a scarcity of neoantigens that can be recognized by immune cells. This classification underscores its low immunogenicity and high immunosuppressive characteristics, which are likely influenced by immunodeficiency and immunosuppression in the tumor microenvironment (TME). Recent comprehensive studies of the TME have provided valuable insights into the pathogenesis of PC and potential targeted therapies. Therefore, understanding the role of each immunophenotype within the PC TME is essential for developing effective therapeutic strategies.
Immune checkpoint inhibition has transformed cancer treatment over the past decade, with nearly 70 different FDA-approved indications spanning more than 18 histologies. However, these therapies have not demonstrated significant clinical benefits in pancreatic cancer (PC), except for a small subset of patients (less than 1%) who exhibit microsatellite instability (MSI) in their tumors [12]. Single-agent therapies and combinations of PD-1, PD-L1, or CTLA-4 inhibitors have proven ineffective in patients with advanced pancreatic cancer, with objective response rates (ORRs) of less than 5% [13],[14],[15], including patients with positive PD-L1 expression, a biomarker that has been associated with improved responses in other cancers. Proposed mechanisms of resistance to immunotherapy in pancreatic cancer (PC) encompass poor T cell infiltration, low tumor mutational burden, and a highly immunosuppressive tumor microenvironment (TME). However, recent comprehensive profiling of PC tumors suggests that 20–30% of patients may exhibit moderate T cell content, and in certain contexts, the presence of tumor immunogenic neo-epitopes and T cell immunity can correlate with overall survival (OS) [16],[17],[18].
The close link between cancer and inflammation, keenly observed by Virchow [19] in the 19th century, foreshadowed contemporary concerns about a possible immunological role in neoplastic pathogenesis. As Harold Dvorak [20] has observed, inflammation bears a striking resemblance to a wound that cannot heal. Currently, an estimated 20% of global cancer mortalities relate to unhealed infections and/or inflammation, and a high proportion of this burden of disease is attributable to gastrointestinal malignancies [21]. Despite these instances of the immune system contributing to tumorigenesis and progression, there is also a wealth of data supporting the protective role of immunity in tumor suppression. While numerous cross-sectional and cohort studies have investigated the relationship between immune cells and PC cancer [22],[23], their observational nature limits them to establishing correlations rather than causations [23]. Although randomized controlled trials (RCTs) could infer causation, interventions to manipulate immune cells are neither feasible nor ethical, thus constraining our ability to draw causal inferences. Given the limited evidence from observational and interventional studies, the Mendelian randomization (MR) approach in human genetics presents a unique opportunity to robustly explore the potential causal links between increased immune cells and PC cancer [24]. This approach leverages the random allocation of genetic variation at conception, well before the onset of disease, making MR a valuable tool for establishing causality and mitigating the risk of reverse causality, independent of confounders typically present in study designs [25],[26],[27].
Here, we utilized MR to investigate the histophysiology and pathophysiological involvement of the immune cells in the development of PC cancer, achieved by a recent statistical summary from a genome-wide association study (GWAS) focused on immune cells [28]. Our study is dedicated to exploring the causal relationship between 731 immune cells and PC cancer, with a special focus on those in tumor initiation, progression, and treatment resistance. We present an extensive MR study that not only identifies specific immune cells associated with PC cancer but also addresses the constraints in current research. Our goal is to provide valuable insights that could refine future immune cell methodologies and advance etiological research. This work is intended to support precision prevention, control, and the development of innovative therapeutic approaches.
MATERIALS AND METHODS
Study design
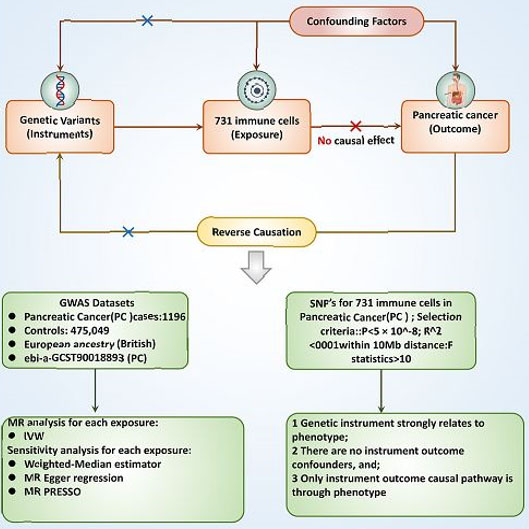
The cause-and-effect relationship between 731 immune cells and PC was assessed using two-sample MR analyses. Mendelian randomization leverages genetic variations as proxies for risk factors. To ensure reliable causal inference, instrumental variables (IVs) used in MR, three key assumptions must be satisfied: (1) The genetic variation must be associated with the exposure directly; (2) The genetic variant is not linked with potential confounding factors between the exposure and outcome; and (3) The genetic variation influences the outcome exclusively through the exposure, not via alternative pathways (Figure 1).
Data sources for exposure and outcome
A summary of Genome-Wide Association Study (GWAS) statistics for each immune trait is publicly accessible from the GWAS catalog (accession numbers: GCST0001391 to GCST0002121) [29]. We used cancer’s keywords to find the immune traits from (https://gwas.mrcieu.ac.uk/). The immune traits included: ebi-a-GCST90018893 (PC). A total of 731 immunophenotypes, including absolute cell (AC) counts, median fluorescence intensities (MFI, which reflect surface antigen levels), morphological parameters (MP) and relative cell (RC) counts, were included. The MFI, AC, and RC features contain B cells, CDCs, mature T cells, monocytes, myeloid cells, TBNK (T cells, B cells, natural killer cells) cells, and Treg panels, while the MP feature contains CDC and TBNK panels. The GWAS database is a comprehensive collection of genetic variation and its association with various traits or diseases. It provides a valuable resource for researchers and clinicians interested in understanding the genetic basis of complex traits and diseases. Based on the inhibitor of differentiation (ID) of cancer, we used online data from GWAS including 476,245 European individuals (n = 1,196 case patients and 475,049 control participants) for PC to analyze the relationship between immune cells and PC according to IDs (immune traits) (https://www.ebi.ac.uk/gwas/).
Instrument selection
Considering that the single-nucleotide polymorphisms (SNPs) number demonstrating genome-wide significance (p < 5 × 10-8) for immune cells traits is extensive, we implemented more stringent correlation thresholds (p < 5 × 10-9) for Genetic Instrumental Variables (IVs) selection [28]. These IVs were identified by grouping them according to the reference panel of the Linkage Disequilibrium (LD) from the 1000 Genomes Project, with a threshold of R2 < 0.001 at a distance of 1,000 kilobases (kb). Given the relatively limited size of the GWAS data for immune cells, we employed a p-value cutoff of 5 × 10-5 and a less significant clustering threshold (R2 < 0.001 at a distance of 1000 kb) [29]. To ensure the reliability of our tools, we selected IVs with F-statistics exceeding 10, identifying them as strong instruments for subsequent analyses. We then extracted these IVs from the summary statistics pertaining to PC outcomes, excluding any that showed potential pleiotropic effects (p < 10-5) on PC, in line with methodologies from previous studies [30]. To maintain consistency in our analysis, we synchronized the SNPs between the exposure and outcome datasets, ensuring uniform effect estimates for the same effect allele. Any alleles with mid-range effect the frequencies of allele (EAFs > 0.42) or SNPs incompatible with the allele were excluded from our analysis [29].
Statistical analysis
In our study, we employed a range of genetic variants as instrumental variables rather than relying solely on an allele score. This approach was chosen to thoroughly examine key assumptions, uncover potential pleiotropy, and facilitate more effective sensitivity and multivariable Mendelian randomization (MR) analyses [25]. To assess the consistency of our findings under different assumptions about heterogeneity and pleiotropy, we utilized four distinct MR methodologies: the inverse variance weighting (IVW; random-effects model), weighted median, MR-Egger, and MR-PRESSO. The IVW method, employing a random-effects model, served as the primary analysis framework for all four sets of instrumental variables. We quantified heterogeneity using Cochran’s Q statistic.
Our study also included analyses with more stringent conditions. The IVW method, under the assumption that all genetic variants are valid, can be biased if many SNPs are influenced by horizontal pleiotropy [31]. Conversely, the weighted median approach, effective when less than 50% of variants exhibit horizontal pleiotropy, presumes most genetic variants are valid [32]. In cases where over 50% of variants are affected by horizontal pleiotropy, we evaluated the strength of our genetic instruments through F-statistics, considering a mean F-statistic of less than 10 indicative of weak instrumental variables [33].
Furthermore, the MR-Egger method was applied to check for potential directional pleiotropy. Here, a significant intercept would indicate a violation of instrumental variable assumptions, suggesting directional pleiotropy [34]. We also implemented the MR pleiotropy residual sum and outlier (PRESSO) method, designed to minimize heterogeneity in causal effect estimates by excluding disproportionately influential SNPs (NbDistribution = 1,500) [35]. Additionally, Steiger-filtering analyses were conducted to detect and eliminate genetic variants more strongly associated with the outcome than the exposure, indicating possible reverse causality [36].
All statistical analyses were performed using R version 4.3.1 (R Foundation) and specific R packages (“TwoSampleMR” and “Mendelian Randomization”) tailored for MR analysis [37],[38]. The TwoSampleMR package provided causal estimates from the four MR models (IVW, weighted median, MR-Egger, and MR-PRESSO), and the MendelianRandomization package was utilized for multivariable MR. Detailed methodologies are further provided in the online Supporting Information Methods.
RESULTS
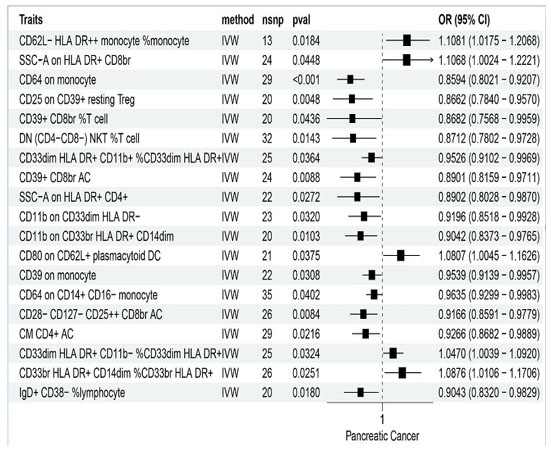
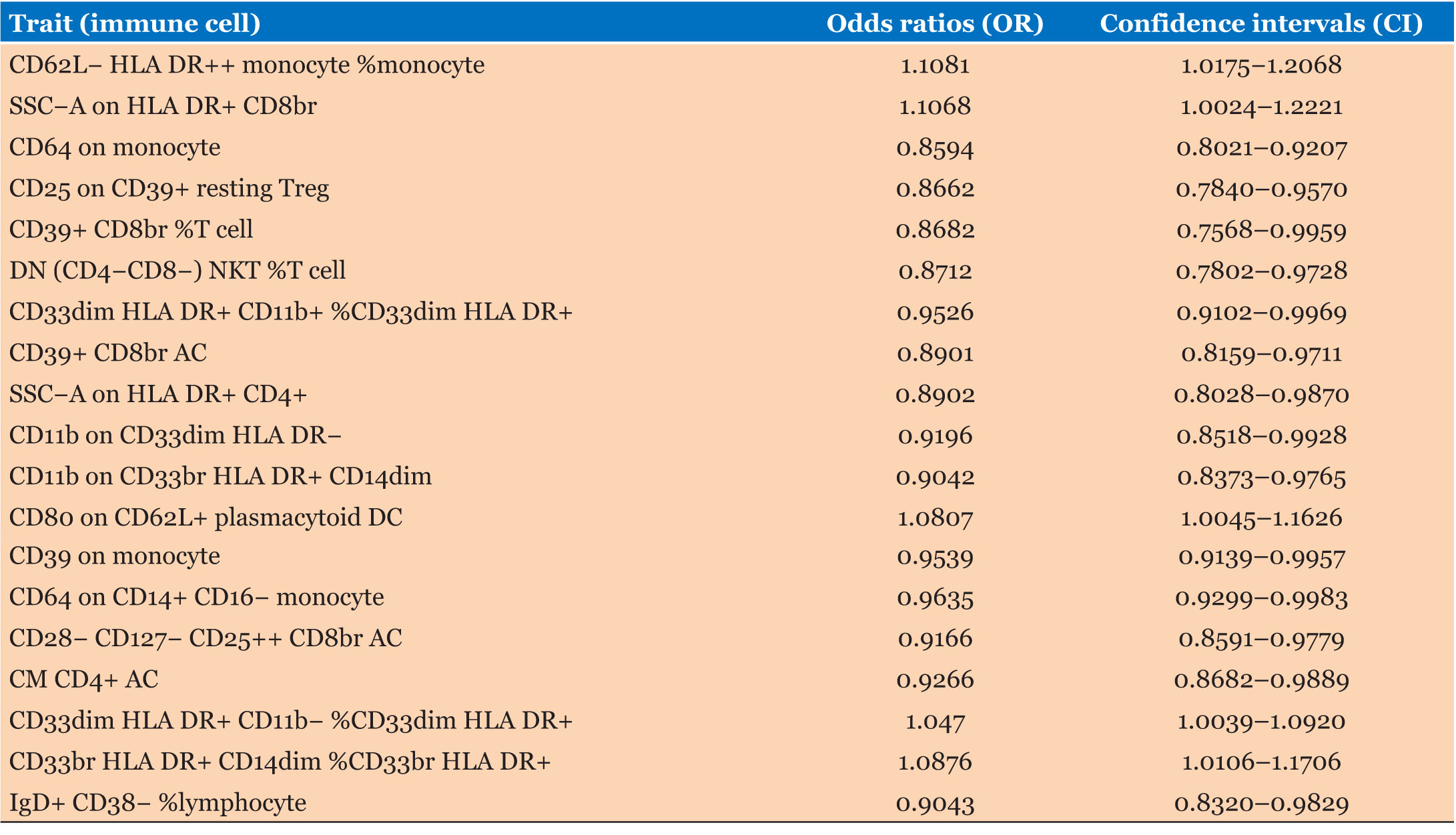
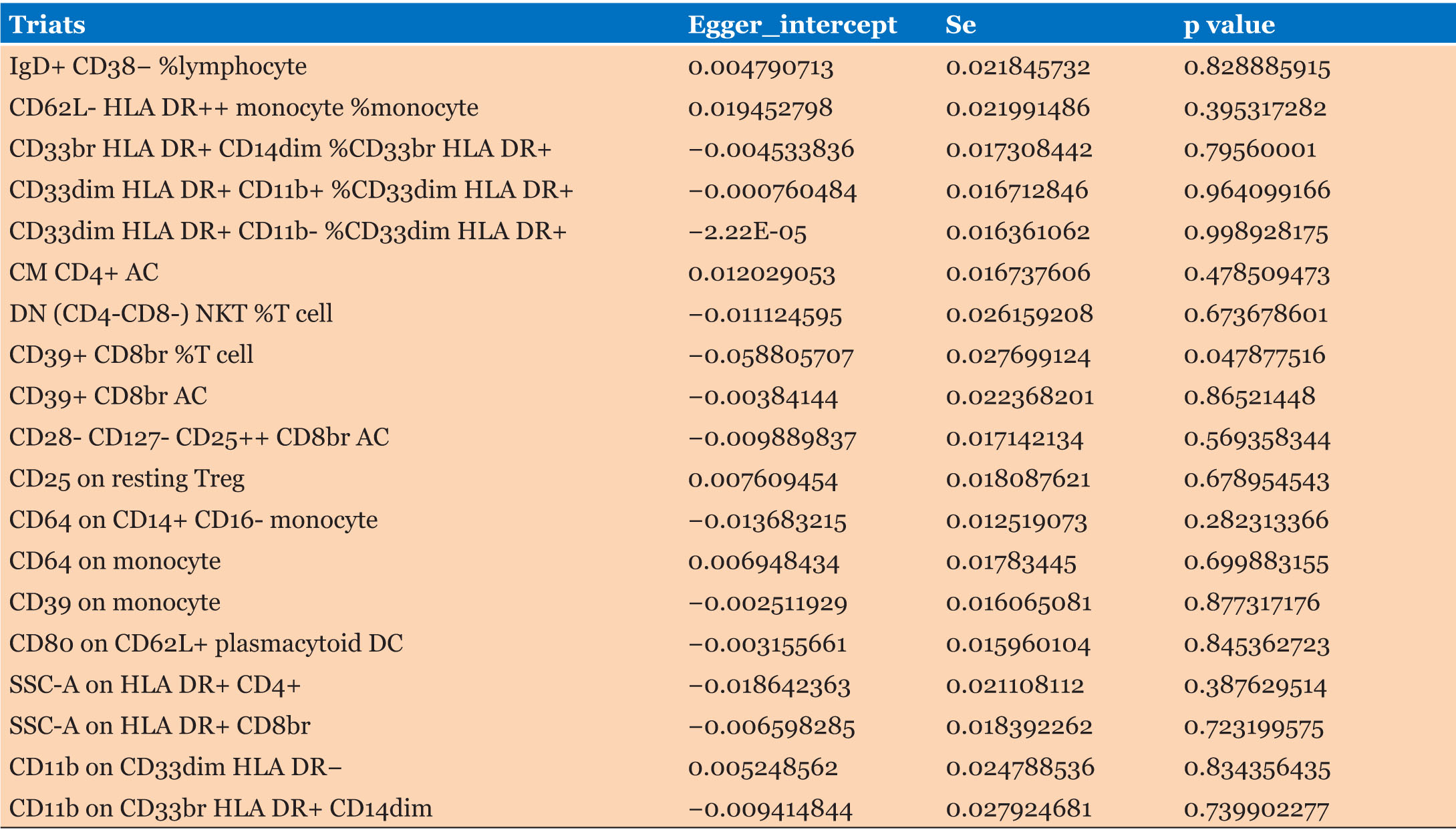
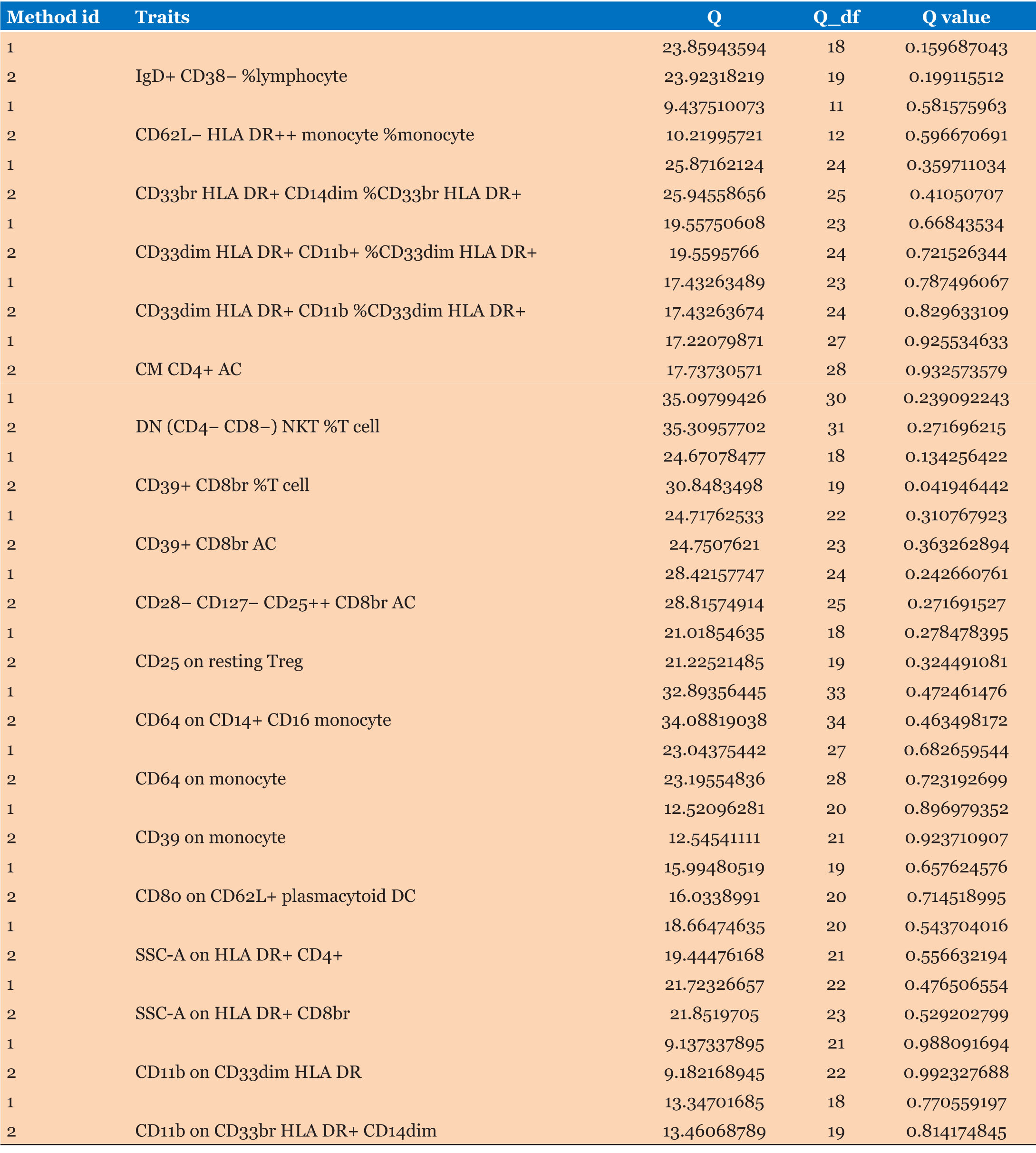
The causal associations between immune cells and PC are summarized in Figure 2 and Table 1. Inverse variance weighting results showed a strong correlation between PC risk and CD62L– HLA DR++ monocyte % monocytes (OR = 1.1081, 95% CI = 1.0175–1.2068, p = 0.0184), SSC–A on HLA DR+ CD8br (OR = 1.1068, 95% CI = 1.0024–1.2221, p = 0.0448), CD64 on monocytes (OR = 0.8594, 95% CI = 0.8021–0.9207, p < 0.001), DN (CD4-CD8–) NKT %T cells (OR = 0.8712, 95% CI = 0.7802–0.9728, p = 0.0143), and SSC–A on HLA DR+ CD4+ cells (OR = 0.8902, 95% CI = 0.8028–0.9870, p = 0.0272). There was a weaker association between PC and CD33dim HLA DR+ CD11b+ % CD33dim HLA DR+ (OR = 0.9526, 95% CI = 0.9102–0.9969, p = 0.0364), CD11b on CD33dim HLA DR– (OR = 0.9196, 95% CI = 0.8518–0.9928, p = 0.0320), CD11b on CD33br HLA DR+ CD14dim (OR = 0.9042, 95% CI = 0.8373–0.9765, p = 0.0103), CD80 on CD62L+ plasmacytoid DCs (OR = 1.0807, 95% CI = 1.0045–1.1626, p = 0.0375), CD64 on CD14+ CD16– monocytes (OR = 0.9635, 95% CI = 0.9299–0.9983, p = 0.0402), CD28– CD127– CD25++ CD8br AC (OR = 0.9166, 95% CI = 0.8591–0.9779, p = 0.0084), CM CD4+ AC (OR = 0.9266, 95% CI = 0.8682–0.9889, p = 0.0216), CD33dim HLA DR+ CD11b– %CD33dim HLA DR+ (OR = 1.0470, 95% CI = 0.8591–0.9779, p = 0.0324), CD33br HLA DR+ CD14dim % CD33br HLA DR+ (OR = 1.0876, 95% CI = 1.0106–1.1706, p = 0.0251), and gD+ CD38–%lymphocytes (OR = 0.9043, 95% CI = 0.8320–0.9829, p = 0.0180). Neither the MR-Egger intercept test nor Cochran’s Q test revealed pleiotropy or heterogeneity (Table 2 and Table 3).
DISCUSSION
Mendelian randomization analysis has been frequently employed to illustrate possible causality between risk factors and diseases. In the present study, we used MR to generate proof of an inverse causal relationship between immune cells and PC. In our study, we found a total of 19 immune cells associated with PC include CD62L-HLA DR++ monocyte % monocytes, SSC–A on HLA DR+ CD8br, CD64 on monocytes, DN (CD4- CD8–) NKT %T cells, SSC–A on HLA DR+ CD4+ cells, CD33dim HLA DR+ CD11b+ % CD33dim HLA DR+, CD11b on CD33dim HLA DR, CD11b on CD33br HLA DR+ CD14dim, CD80 on CD62L+ plasmacytoid DCs, CD64 on CD14+ CD16-monocytes, CD28– CD127– CD25++ CD8br AC, CM CD4+ AC, CD33dim HLA DR+ CD11b– % CD33dim HLA DR+, CD33br HLA DR+ CD14dim % CD33br HLA DR+, and gD+ CD38– %lymphocytes. Among them, CD62L–HLA DR++ monocyte % monocytes, SSC–A on HLA DR+ CD8br, CD80 on CD62L+ plasmacytoid DCs, CD33dim HLA DR+ CD11b– % CD33dim HLA DR+, CD33br HLA DR+ CD14dim % CD33br HLA DR+ are risk factors for PC, while the rest of immune cells are protective factors for PC. We have highlighted some of the strongest associations in the following discussion sections.
We found that CD62L-HLA DR++ monocyte infiltration was closely related to PC, and was a negative factor for PC prognosis. These monocytes express HLA-DR strongly but do not express CD62L. HLA-DR is a major histocompatibility complex (MHC) class II cell surface marker that is regulated by the human leukocyte antigen (HLA) complex located on chromosome 6 (region 6P21), and it contains two subunits with molecular weights of 36 and 27 kD, respectively (α Subunit and β Subunits). It is expressed mainly on antigen-presenting cells. Previous research has suggested that tumor-specific MHC-II expression is associated with good outcomes. For example, upregulated HLA-DR/MHC-II genes in tumors serve as predictive factors for neoadjuvant chemotherapy responses and good clinical outcomes in locally advanced rectal cancer [39]. In addition, increased MHC-II expression in tumor cells is associated with improved melanoma treatment responses, progression-free survival (PFS), and OS [40]. It can also be used as a positive predictor of good outcomes in Hodgkin lymphoma after PD-1 blockade [41]. Interestingly, HLA-DR expression in non-tumor cells cannot predict treatment responses. CD62L is a gene encoding L-selectin protein, which belongs to the selectin family. Selectin is a cellular adhesion molecule related to leukocyte adhesion and migration. In some tumors, the expression level of tumor cell surface selectin significantly increases. In melanoma, increases in tumor-infiltrating CD62L+T cells promote growth, suggesting that CD62L is associated with poor prognosis, consistent with our present findings. We found that increases in CD62L-HLA DR++ monocyte infiltration were a risk factor for the development of PC.
SSC–A on HLA DR+ CD8br is a late-activated CD8+T cell. Overexpression of HLA DR+CD8br cells in acute myeloid leukemia may weaken the anti-tumor efficacy and benefits of relapsed refractory acute myeloid leukemia, which may be related to T cell depletion [42]. Interestingly, we found that activation of SSC–A on HLA DR+ CD8br was beneficial to PC prognosis, but the specific mechanism still needs further research. Another immune cell CD64 on monocytes, in humans, CD64 on monocytes are IgG Fc receptors (Fc γ R). There are three main categories of these receptors. Polymorpho nuclear phagocytes typically express the low-affinity receptor category (Fc γ RH [CD32] and FC γ RIII [CD16]). Studies have shown that, in malignant tumors, neutrophils overexpress high-affinity CD64 Fc receptors after granulocyte colony-stimulating factor (G-CSF) treatment γ RI [43]. In addition, CD64 is closely related to cancer immunotherapy [44]. We observed that monocyte CD64 overexpression was a protective factor for PC, but the mechanism underlying this interaction remains unclear.
Double-negative (CD4- CD8–) NKT %T cells account for a small proportion of circulating T lymphocytes, with phenotypic features such as loss of CD4 and CD8 co-receptors and γδ or αβ. T cell receptors (TCR) have complex roles, and the exclusion of skin and cardiac allotransplantation by exclusively suppressing anti-transplant-specific CD8+ T cell functions can have negative impacts [45]. Inhibiting DN (CD4- CD8) T cell activation can reduce IFN-γ-mediated inflammatory responses. However, a previous study found increases in the proportion of DN (CD4- CD8-) T cells in thyroid cancer, indicating that tumor growth may be related to DN (CD4- CD8-) T cell infiltration. DN T cells can express perforin and granzymes, which kill NK cells in the tumor microenvironment [46]. Double-negative T cells may also block NK-mediate pro-inflammatory immune environments and promote cancer cell survival [47]. Interestingly, recent studies have shown that DN (CD4- CD8-) tumor-infiltrating lymphocytes derived from solid tumor tissue inhibit tumor cell proliferation in an MHC-independent manner after expansion in vitro. Our study showed that DN (CD4- CD8-) NKT %T cells activation was a protective factor for PC.
Amplification of myeloid-derived suppressor cells (MDSCs) is associated with tumorigenesis in colorectal cancer (CRC). Here, we identified a strong correlation between CD33+ MDSC levels and the levels of Yes-associated protein 1 (YAP1) and phosphatase and tensin homolog (PTEN) in CRC patients. Tumor expression of YAP1 and PTEN is correlated with the amplification of tumor-related myeloid-derived suppressor cells and decreased CRC survival [48]. Granulocyte MDSCs express CD33, CD11b, IL-4Ra, and low levels of CD15 and denote elevated levels of arginase. Myeloid-derived suppressor cell levels were notably increased in esophageal cancer (EC), gastric cancer (GC), and PC in our study, and increasing MDSC percentages were significantly correlated with an increased risk of mortality in previous work [49]. Another study demonstrated that high Birc5 expression and high MDSC infiltration within tumors were associated with HCC patients’ prognosis. In vitro, hepatocyte Birc5 overexpression facilitated the expansion of immunosuppressive CD11b+CD33+HLADR-MDSCs in human peripheral blood mononuclear cells. Transgenic animal models of HCC showed that Birc5 deficiency upregulated genes were implicated in lymphocyte-mediated immunity, natural killer cell-mediated immunity, gamma-interferon production, T-cell activation, and T-cell-mediated cell cytotoxicity, which is in line with our finding of a correlation between CD33 and HCC risk [50]. These findings were consistent with ours in that CD33+ MDSC levels were associated with poor prognosis in PC. We also found that there were two CD33 subtypes: CD33dim HLA DR+ CD11b- %CD33dim HLA DR+ and CD33dim HLA DR+ CD11b+ %CD33dim HLA DR+. We found that CD33dim HLA DR+ CD11b-%CD33dim HLA DR+ was more commonly a risk factor for cancer development, whereas CD33dim HLA DR+ CD11b+ %CD33dim HLA DR+ was a protective factor.
Interestingly, CM CD4+ AC typically represents a distinct subpopulation of immune cells characterized by the presence of CD4 co-receptors. CD4 is a co-receptor primarily found on helper T cells and is important for the interaction between T cells and antigen-presenting cells. Analysis of CD4+ cells may be important for both research and clinical purposes. CD4+ T cells are key coordinators of the immune system, as they produce several cytokines after activation and differentiation. CD4+ T helper cell subtypes (including T helper 1, T helper 2, T helper 17, T helper 9, and regulatory T cells) have different immune functions after differentiation from naïve T cells. Different types of CD4+ T cells require different cytokines and master transcription factors for activation [51]. Previous research has demonstrated that CD4+ T cells can be found in the tumor microenvironments of lung cancer, melanoma, CRC, lymphomas, cervical cancer, and ovarian cancer, but the role of CD4+ T cells in EC is relatively understudied [52],[53],[54],[55],[56],[57]. Interestingly, our findings indicated that the presence of CM CD4+ T cells was a protective factor in PC. More functional research is needed to confirm these findings.
Strength and limitations
This study employed published GWAS data to perform a two-sample Mendelian randomization (MR) analysis, benefiting from a large sample size and strong statistical power. The findings are based on genetic instrumental variables, with causal inferences supported by various MR methodologies. The results are robust, demonstrating resilience against confounding factors, including horizontal pleiotropy. This approach addresses limitations of traditional observational studies by minimizing the impact of confounding variables and reverse causality. Additionally, MR helps overcome issues of representativeness and feasibility that are often associated with randomized controlled trials (RCTs). However, it is important to note a limitation of the study: its reliance on GWAS databases raises concerns about the applicability of the findings to the immune cell profile in pancreatic cancer (PC). Further research and validation across diverse populations are warranted, as the results are primarily generalizable to European populations due to the demographic focus of the original GWAS. Additional studies are needed to confirm these findings in non-European populations.
CONCLUSION
Our comprehensive two-sample Mendelian randomization (MR) analysis has revealed a causal connection between 19 different immune cell phenotypes and pancreatic cancer (PC), including specific immune cells such as monocytes, T cells, lymphocytes, and dendritic cells (DCs). This finding highlights the complex relationship between the immune system and PC, paving the way for further exploration of the underlying biological mechanisms and potential immunotherapeutic strategies for this cancer.
REFERENCE
1.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34. [CrossRef]
[Pubmed]

2.
Klein AP. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol 2021;18(7):493–502. [CrossRef]
[Pubmed]
3.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68(6):394–424. [CrossRef]
[Pubmed]
4.
Doll R, Peto R, Boreham J, Sutherland I. Mortality from cancer in relation to smoking: 50 years observations on British doctors. Br J Cancer 2005;92(3):426–9. [CrossRef]
[Pubmed]
5.
Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156(19):2255–60.
[Pubmed]
6.
Larsson SC, Permert J, Håkansson N, Näslund I, Bergkvist L, Wolk A. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. Br J Cancer 2005;93(11):1310–5. [CrossRef]
[Pubmed]
7.
Lynch SM, Vrieling A, Lubin JH, et al. Cigarette smoking and pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Am J Epidemiol 2009;170(4):403–13. [CrossRef]
[Pubmed]
8.
Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22(44):9694–705. [CrossRef]
[Pubmed]
9.
Mathers CD, Fat DM, Inoue M, Rao C, Lopez AD. Counting the dead and what they died from: An assessment of the global status of cause of death data. Bull World Health Organ 2005;83(3):171–7.
[Pubmed]
10.
Liu L, Zhao G, Wu W, et al. Low intratumoral regulatory T cells and high peritumoral CD8(+) T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunol Immunother 2016;65(1):73–82. [CrossRef]
[Pubmed]
11.
Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4(6):437–50. [CrossRef]
[Pubmed]
12.
Sharma P, Siddiqui BA, Anandhan S, et al. The next decade of immune checkpoint therapy. Cancer Discov 2021;11(4):838–57. [CrossRef]
[Pubmed]
13.
Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65. [CrossRef]
[Pubmed]
14.
Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21(19):4286–93. [CrossRef]
[Pubmed]
15.
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33(8):828–33. [CrossRef]
[Pubmed]
16.
Balachandran VP, łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551(7681):512–6. [CrossRef]
[Pubmed]
17.
Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res 2017;23(12):3129–38. [CrossRef]
[Pubmed]
18.
Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res 2017;5(11):978–91. [CrossRef]
[Pubmed]
19.
Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow? Lancet 2001;357(9255):539–45. [CrossRef]
[Pubmed]
20.
Dvorak HF. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986;315(26):1650–9. [CrossRef]
[Pubmed]
21.
Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990;98(3):694–702. [CrossRef]
[Pubmed]
22.
Schlanger D, Popa C, PaÛca S, Seicean A, Al Hajjar N. The role of systemic immuno-inflammatory factors in resectable pancreatic adenocarcinoma: A cohort retrospective study. World J Surg Oncol 2022;20(1):144. [CrossRef]
[Pubmed]
23.
Yamaguchi Y, Katata Y, Okawaki M, Sawaki A, Yamamura M. Republication: A prospective observational study of adoptive immunotherapy for cancer using zoledronate-activated killer (ZAK) cells – An analysis for patients with incurable pancreatic cancer. Anticancer Res 2022;42(2):1181–7. [CrossRef]
[Pubmed]
24.
Pingault JB, O’Reilly PF, Schoeler T, Ploubidis GB, Rijsdijk F, Dudbridge F. Using genetic data to strengthen causal inference in observational research. Nat Rev Genet 2018;19(9):566–80. [CrossRef]
[Pubmed]
25.
Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [CrossRef]
[Pubmed]
26.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA 2017;318(19):1925–6. [CrossRef]
[Pubmed]
27.
Smith GD, Ebrahim S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32(1):1–22. [CrossRef]
[Pubmed]
28.
Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet 2020;52(10):1036–45. [CrossRef]
[Pubmed]
29.
Cai J, Li X, Wu S, et al. Assessing the causal association between human blood metabolites and the risk of epilepsy. J Transl Med 2022;20(1):437. [CrossRef]
[Pubmed]
30.
Zeng P, Wang T, Zheng J, Zhou X. Causal association of type 2 diabetes with amyotrophic lateral sclerosis: New evidence from Mendelian randomization using GWAS summary statistics. BMC Med 2019;17(1):225. [CrossRef]
[Pubmed]
31.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017;46(6):1985–98. [CrossRef]
[Pubmed]
32.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40(4):304–14. [CrossRef]
[Pubmed]
33.
Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir Med 2017;5(11):869–80. [CrossRef]
[Pubmed]
34.
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 2017;28(1):30–42. [CrossRef]
[Pubmed]
35.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50(5):693–8. [CrossRef]
[Pubmed]
36.
Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 2017;13(11):e1007081. [CrossRef]
[Pubmed]
37.
Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [CrossRef]
[Pubmed]
38.
Yavorska OO, Burgess S. MendelianRandomization: An R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46(6):1734–9 [CrossRef]
[Pubmed]
39.
Zhang S, Li N, Wang F, et al. Characterization of the tumor microenvironment and identification of spatially predictive biomarkers associated with beneficial neoadjuvant chemoradiotherapy in locally advanced rectal cancer. Pharmacol Res 2023;197:106974. [CrossRef]
[Pubmed]
40.
Johnson DB, Estrada MV, Salgado R, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016;7:10582. [CrossRef]
[Pubmed]
41.
Roemer MGM, Redd RA, Cader FZ, et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol 2018;36(10):942–50. [CrossRef]
[Pubmed]
42.
Fang J, Zhang R, Lin X, Xu Y, Huang K, Saw PE. Aberrant expression of T cell activation markers and upregulation of Tregs in bone marrow and peripheral blood in acute myeloid leukemia patients. Hematology 2023;28(1):2219554. [CrossRef]
[Pubmed]
43.
Hui GK, Gao X, Gor J, Lu J, Sun PD, Perkins SJ. The solution structure of the unbound IgG Fc receptor CD64 resembles its crystal structure: Implications for function. PLoS One 2023;18(9):e0288351. [CrossRef]
[Pubmed]
44.
Valerius T, Repp R, de Wit TP, et al. Involvement of the high-affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood 1993;82(3):931–9.
[Pubmed]
45.
Fischer K, Voelkl S, Heymann J, et al. Isolation and characterization of human antigen-specific TCR alpha beta+ CD4(–)CD8– double-negative regulatory T cells. Blood 2005;105(7):2828–35. [CrossRef]
[Pubmed]
46.
He KM, Ma Y, Wang S, et al. Donor double-negative Treg promote allogeneic mixed chimerism and tolerance. Eur J Immunol 2007;37(12):3455–66. [CrossRef]
[Pubmed]
47.
Imam S, Dar P, Paparodis R, et al. Nature of coexisting thyroid autoimmune disease determines success or failure of tumor immunity in thyroid cancer. J Immunother Cancer 2019;7(1):3. [CrossRef]
[Pubmed]
48.
Yang R, Cai TT, Wu XJ, et al. Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology 2018;155(2):263–72. [CrossRef]
[Pubmed]
49.
Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother 2011;60(10):1419–30. [CrossRef]
[Pubmed]
50.
Liu Y, Chen X, Luo W, et al. Identification and validation of Birc5 as a novel activated cell cycle program biomarker associated with infiltration of immunosuppressive myeloid-derived suppressor cells in hepatocellular carcinoma. Cancer Med 2023;12(15):16370–85. [CrossRef]
[Pubmed]
51.
Ashfaq H, Soliman H, Saleh M, El-Matbouli M. CD4: A vital player in the teleost fish immune system. Vet Res 2019;50(1):1. [CrossRef]
[Pubmed]
52.
Kong CY, Sigel K, Criss SD, et al. Benefits and harms of lung cancer screening in HIV-infected individuals with CD4+ cell count at least 500 cells/µl. AIDS 2018;32(10):1333–42. [CrossRef]
[Pubmed]
53.
Niakosari F, Sur M. Agranular CD4+/CD56+ hematodermic neoplasm: A distinct entity described in the recent World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas. Arch Pathol Lab Med 2007;131(1):149–51. [CrossRef]
[Pubmed]
54.
Oliveira G, Stromhaug K, Cieri N, et al. Landscape of helper and regulatory antitumour CD4+ T cells in melanoma. Nature 2022;605(7910):532–8. [CrossRef]
[Pubmed]
55.
Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune checkpoints in circulating and tumor-infiltrating CD4+ T cell subsets in colorectal cancer patients. Front Immunol 2019;10:2936. [CrossRef]
[Pubmed]
56.
Ukita M, Hamanishi J, Yoshitomi H, et al. CXCL13- producing CD4+ T cells accumulate in the early phase of tertiary lymphoid structures in ovarian cancer. JCI Insight 2022;7(12):e157215. [CrossRef]
[Pubmed]
57.
Li R, Liu Y, Yin R, et al. The dynamic alternation of local and systemic tumor immune microenvironment during concurrent chemoradiotherapy of cervical cancer: A prospective clinical trial. Int J Radiat Oncol Biol Phys 2021;110(5):1432–41. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Author Contributions
Pengkhun Nov - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Socheat Touch - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Syphanna Sou - Conception of the work, Design of the work, Analysis of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Samnang Kouy - Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Duanyu Wang - Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Jiqiang Li - Conception of the work, Design of the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guarantor of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2025 Pengkhun Nov et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.



